AlphaFold-NMR
Scripts and data corresponding to Huang, Ramelot, Spaman, Kobayashi, Montelione (2025) "Hidden Structural States of Proteins Revealed by Conformer Selection with AlphaFold-NMR"
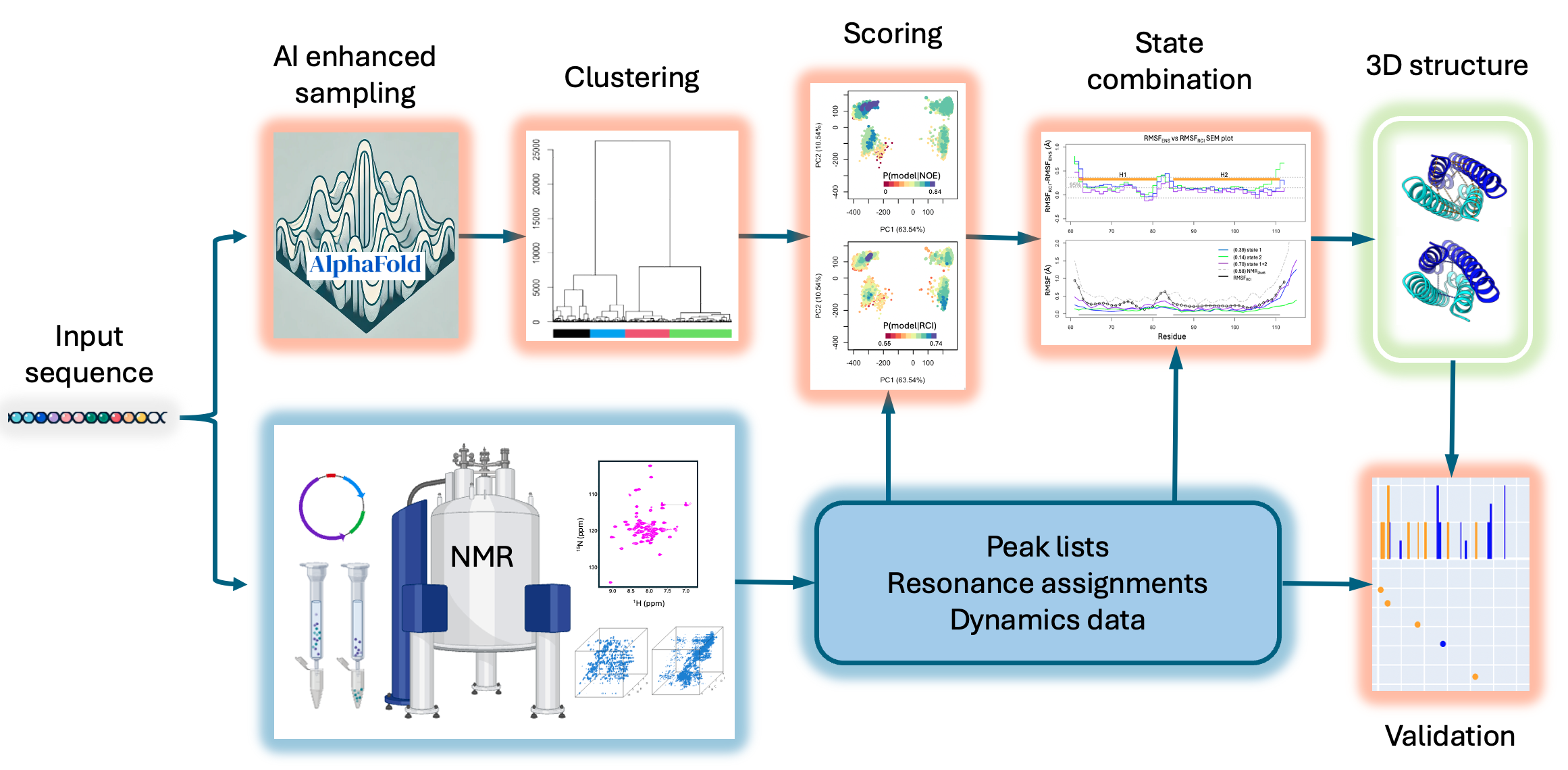
Usage
Data and scripts for CDK2AP1-doc1 AF-NMR analysis:
- AI enhanced sampling: CDK2AP1-doc1/EnhancedSampling/
- doc1_noN.fasta: input fasta sequence. We exclude the long disordered tails and non-native tags from the input fasta sequence for AF modeling, to avoid potential influence on the pTM and <pLDDT> scores.
Commands:
sbatch run_doc1_noN.sh (running with slrum)
This command calculates and relax all 6000 models using run_afsample6000.sh
The output models are here: AF_models_dropout/doc1_noN. pTM score is reported here: AF_models_dropout/scores.sc and log from AF: slurm-xxx.out
python FilterAF2.py -log slurm-xxx.out -rel -inD AF_models_dropout/doc1_noN -outD filteredModels
This command filters out bad models based on the AF log file (e.g. slurm-xxx.out). The python code is copied from here: https://github.rpi.edu/RPIBioinformatics/FilteringAF2_scripts
Additional processing scripts:
Merge two chains into one chain for clustering analysis
sh runMergedChain.py filteredModels mergedModels
This command finds all pdb file in the fileredModels, merge two chains (using mergeChain.py) and save them in the mergedModels directory.
5984 models with one merged chain (CDK2AP1-doc1/ESmodels/) are used for the following analyses:
- Clustering
R scripts for CDK2AP1:
CDK2AP1-doc1/Clustering/: dmPCAClustering.R --> output: pc_dm_pdbs.RData, cluster_pc_dm.csv (in Rstudio, set the working dir to this dir before running the R script)
We found that "ward methods" gives largest agglomerative coefficient. Number of clusters --> by viusal inspection of "Dendrogram" and pc plots to identify number of well-seperated clusters.
- Scoring
NMRdata:
- CDK2AP1-doc1/RPF/: directory to calculate RPF scores:
- Input/
- CDK2AP1-doc1/RCI/: directory to calculate RCI and SCC scores
- RPFtable2SHIFTY.py: convert the chemical shift file used by RPF (bmrbtable file) to SHIFTY format
- nmrstar3toSHIFTY-fromBMRB.py: give the bmrb ID number, download chemical shift assignments from the BMRB database and convert to SHIFTY format
- nmrstar3toSHIFTY.py: convert the local bmrb file in nmrstart 3.0 format to SHIFTY format
Other scores:
- CDK2AP1-doc1/Scoring/:
- combine all scores:
- plots:
- State combination
scripts for CDK2AP1:
- Validation
scripts for CDK2AP1: